



昨天的文章《连花清瘟能治疗新冠,国际医学界承认吗?》发出后,评论区和私信中有网友问我,除了连花清瘟,还有哪些中成药曾经申请过FDA?
尽管有人会表示不屑:中医药和西医药的用药原理和使用方法截然不同,作为经历了几千年验证的国之瑰宝,中药凭什么要用西医的标准接受FDA的评判?
但从国内部分中医药企业的行为来看,他们本身并不认可这种思维。
今天就让我来盘点一下,除了连花清瘟之外,还有哪些中成药曾经冲击过FDA。
根据2017年版的《中国医药产业国际化蓝皮书》,迄今为止,国内有10款中药向FDA提出过申请。
这些企业和药物分别是:
- 天士力制药集团股份有限公司的复方丹参滴丸
- 和记黄埔医药有限公司的HMPL-004
- 北京北大维信生物科技有限公司的血脂康胶囊
- 上海现代中医药股份有限公司的扶正化瘀片
- 江苏康缘药业股份有限公司的桂枝茯苓胶囊
- 上海杏灵科技药业股份有限公司杏灵颗粒
- 华颐药业有限公司的威麦宁胶囊
- 浙江康莱特药业有限公司的康莱特注射液和康莱特软胶囊(两款药物)
- 以岭药业股份有限公司连花清瘟胶囊
显然,上述这些中医药企业并不像他们的支持者那样,对FDA不屑一顾,相反,他们愿意花费大量的时间和金钱去进行尝试。
原因何在呢?
首先,获得FDA的认可进而实现国际化,是国家部委级的战略方针。
早在1996年,国家科委(现科技部)就提出了“敲开FDA大门”的口号,1999年,科技部通过公开征集遴选,从我们号称博大精深的中医药宝库中,精心挑选了复方丹参滴丸、双黄连片、桂枝茯苓丸、七叶皂甙等7个中成药,奔赴美国,向FDA提出申报。值得一提的是,国家科委给每个品种补贴30万元左右,这在当时可不是一个小数目。
我们都听说过我国在体育方面的“奥运战略”,为了提升国民的民族自信心,特意在“小、巧、难、女、少”这几个方面,寻找一些冷门、投入小、容易快速出成绩的项目,来达到尽可能多获得金牌的目的。
同样的,国家科委的“FDA战略”,其目的不仅仅是为了把中医药推向美国市场,更重要的目的也是为了提升国民的民族自信心,为中医药在世界主流医学界,挣回一些颜面。
那这两项战略的结果如何呢?
从最近几届奥运会来看,“奥运战略”确实给我们争取了很多的金牌,让我们在奥运金牌榜常年高居前三。
美中不足的是,尽管一些项目在不断地摘金夺银,而另外一些拥有广大群众基础的项目,例如男子三大球,反而与世界一流水准渐行渐远。
无论如何,奥运战略好歹赢了面子,实实在在地提升了我们的民族自信心。
“FDA战略”表现如何呢?
这里按下不表,先说第二个原因——
叩开FDA的大门,是中医药企业开拓美国甚至全球医药市场的必由之路,简单说,要想到国际上挣大钱,需要先过FDA这关。
FDA,全称美国食品药品监督管理局,是美国卫生与公众服务部直辖的联邦政府机构,其主要职能为负责对美国国内生产及进口的食品、膳食补充剂、药品、疫苗、生物医药制剂、血液制剂、医疗设备、放射性设备、兽药和化妆品进行监督管理。
任何一种药物,如果不能获得FDA的许可,是没有可能作为药物在美国市场进行销售的。
我们知道,不仅美国有食品和药品管理局,世界上每个主要国家都有自己的食品药品管理机构,例如中国的SFDA(中华人民共和国国家食品和药品监督管理局),同样,英国(MHRA)、法国、德国、日本等等,也都有各自的食品药品管理局(或由卫生部管理)。从理论上说,要想进入每个国家的市场,都应该获得该国管理部门的许可。
但是,鉴于FDA是全世界科学性最权威、水平最高、审核及管理最严格的食品及药品管理机构,所以一旦某产品获得了FDA的批准,就能极大地给该产品进入其他国家的市场获得审批加分项。尤其是一些本国管理水平较差的国家,几乎以FDA为唯一参考标准。
所以,获得FDA的批准可以看作除极少数国家(如北朝鲜)之外是全球通行证。
那么,FDA的这套审批流程是如何运作的呢?
关于这个流程,FDA官网有一套比较系统的说明:Development & Approval Process | Drugs


整个流程一共分为12个步骤,我这里结合资料把这12个步骤说清楚:
第一步、临床前研究
1. 药物研发:包括药物的实验室研究以及生物学筛选
这个流程,化学药通常需要2-10年的时间,而中成药则相对较快,例如著名的连花清瘟,大约15天就完成了药物研发。
2. 临床前实验:包括动物实验、药理学研究和毒理学研究。
这个流程化学药通常持续3-6年,而中成药因为分子结构复杂,药理学、毒理学研究通常是一团乱麻。
第二步、研究性新药申请
Investigational New Drug Application,简称IND申请
当一个化合物通过了临床前研究后,就可以向FDA提交IND申请了,以展开后续的人体试验。
如果在提交申请后30天内FDA没有驳回申请,那么该新药临床研究申请即被视为有效,可以进行人体试验。
新药临床研究申请需要提供先前试验的材料;以及计划将在什么地方,由谁以及如何进行临床试验的说明;新化合物的结构;投药方式;动物试验中发现的所有毒性情况;该化合物的制造生产情况。
所有临床方案必须经过机构审评委员会(Institutional Revuew Board,IRB)的审查和通过。
每年必须向FDA和IRB 汇报一次临床试验的进程和结果。
第三步、Ⅰ期临床试验
通常需要征集20-80名正常且健康的志愿者进行试验研究。
该阶段试验的主要目的是进行药物的安全性评价,包括该药物的安全剂量范围。同时也要通过这一阶段的临床试验获得其吸收、分布、代谢和排泄以及药效持续时间的数据和资料。耗时一年左右。
第四步、II期临床试验
通常需要征集100-300名相关病人进行试验研究。
该阶段试验的主要目的是对药物治疗进行初步的有效性评价。通常耗时1-2年。
第五步、III期临床试验
通常需1000-5000名临床和住院病人,要求在多个医学中心进行,在医生的严格监控下,进一步对药物的安全性有效性进行评价,以及与其他药物的相互作用关系。
该阶段试验一般采取多中心,安慰剂对照和双盲法试验。通常耗时1-4年。
第三期临床试验是整个临床试验中最主要的一步。
注:在完成所有三个阶段的临床试验并分析所有资料及数据,如证明该药物的安全性和有效性后,就可以向 FDA提交新药申请了。新药申请需要提供所有收集到的科学资料。通常一份新药申请材料可多达100000页甚至更多。按照法规,FDA应在6个月内审评完新药申请。但是由于大部分申请材料过多,而且有许多不规范,因此往往不能在这么短的时间内完成。
第六步、审查会议
FDA会和新药申请厂家开会,厂家准备提交新药申请。
第七步、新药申请,New Drug Application,简称NDA
在这个过程中,厂家需要向FDA提供所有包括动物实验、临床试验数据和信息,以及药物对人体的作用以及药物的制造工艺。
第八步-第九步、申请复审
NDA被接收后,FDA有60天的时间决定是否归档,在此期间,NDA要被重审。如果FDA觉得厂家提供的申请资料不完整,会拒绝归档。
第十步、药物标签检查
检查药物的标签是否存在误导性信息。
第十一步、生产设备检查
检查药物的生产设备是否准备完善。
第十二步、FDA通过药物申请
NDA申请被FDA同意后,该药物生就获得了在美国上市的资格,供医生和病人选择。但是还必须定期向FDA呈交有关资料,包括该药物的副作用情况和质量管理记录。对于有些药物FDA还会要求做第四期临床试验,以观测其长期副作用情况。
整套流程走完,一款新药从研发到上市,通常需要耗费10亿以上的美金,和十年以上的时间。
有人问,这样做是不是太死板、太教条了?有必要耗费如此巨大的时间、金钱和人力成本来开发一款新药吗?
这是个好问题。
容我反问:如果不花费巨额的资金进行病理学、毒理学研究,如果不招募成百上千的实验对象反复实验,如果不依靠经年累月的长时间观察和数据分析,药物的疗效如何得到保证?药物的长期副作用如何能够被知晓?
我们当然可以减少药物临床试验的实验对象,也可以缩短实验所耗费的时间,而代价,则可能是千千万万人的健康甚至生命。
孰轻?孰重?
即便FDA遵循如此严格的管理标准,也免不了还是有一些药物,在上市后出现了不良反应超过预期的负面作用。
一旦降低标准,结果可想而知。
FDA这个严格的、不近人情的准入门槛,让那些最早从上世纪九十年代开始觊觎欧美市场的中药企业,迄今为止,没有一家能够获取药物上市的资格。
有人说,你说的不对,有些药物,例如血脂康,明明可以在美国销售啊?
这种说法是对美国的医药市场不了解,以血脂康为代表的一部分中药,是没有资格以药物的身份进入美国的,只能作为膳食补充剂或保健品在美国市场上销售。
在上世纪九十年代中期之前,FDA对所有药物,奉行两个“凡是”的政策:
第一,凡是化学结构不清楚的药物,不予受理。
第二,凡是作用机理不明确的药物,不予受理。
我们知道,中药的药物成分极为复杂,一味单药就含有多种活性成分、且多是大分子物质,至于复方药的复杂性则更是多了几个数量级。要把其化学结构搞清楚,难于上青天(我们现在连月球都能上去了,却依然搞不清楚绝大多数中药的化学结构)。既然连化学机构都搞不清楚,弄明白药物的作用机理就更是痴人说梦了。
这两个凡是,简直就是中药过滤器。
那么,是FDA故意刁难中药吗?
并没有,FDA绝不是对中药才有这样严格的要求的,事实上,对于所有准备在美国上市的药物来说,FDA严格的审核,都无异于一场残酷的试炼。
FDA不但没有难为中药,甚至对中草药实行了另外一套较为宽松的标准。
为了方便以植物药为主的中药通过审核,FDA在2004年公布了《植物药研制指导原则》,专门为植物药设定了大量与化学药不同的审核标准。

按照新的标准,提出申请的中药草,化学结构搞不清楚,没关系,药物作用机理弄不明白,也没关系。
只要在一系列临床试验中,能证明你所申报的药物,具有你所宣称的实际疗效,就允许进入美国市场,这样总可以了吧?
FDA甚至把复方丹参滴丸的第一期临床试验都给免了,直接从二期开始。
结果,还是没有一款药物,能在三期临床试验中表现合格。
这就很尴尬了。
很多人不明白,为什么在中国接受过数百甚至上千年“效果验证”的传统中药,要去远渡重洋接受他国的检验?
作为一门“经验科学”,中医药自古传承至今,经历万千古人的使用,难道还不足以说明其疗效吗?
不能,所谓“经验”这玩意儿,有时候是会骗人的,在冰冷理性的科学面前,常常是经不起推敲的,尤其是带有强烈个人意识的“药物疗效”。
而造成“药物疗效”出现误区的主要因素,包括“幸存者偏差”和“安慰剂效应”。
先说幸存者偏差
二战期间,英美对返航的战斗机进行弹痕分析,发现弹痕主要集中在机翼部位,而驾驶舱和油箱很少有中弹痕迹。为了加强飞机的防御机制,他们根据分布在战斗机上的弹孔,决定加固机翼装甲。
结果,飞机的战毁率没有任何的下降。
为什么?
因为那些被击中驾驶舱、邮箱的飞机,根本就飞不回来!
而中弹最多的机翼,恰恰是最不需要被加强的部位。
这就是幸存者偏差。
一直有人认为,中华民族的繁衍至今,最大的功臣是中医药。
从物种延续的角度来看,没有任何一种疾病,可以完全消灭一个生物物种。再强的疾病,也总有其束手无策的对象。恐怖如当年差点灭绝印第安人的天花,以及数十年来让人闻之色变的艾滋病,总会有一部分人,对其具有免疫效果。
但古代的人们并不了解免疫力,也不懂什么是幸存者偏差,于是那些在瘟疫下存活下来的幸存者,免不了会把他们的生还归功于“吃了某种药物”,甚至是“进行了某种法术仪式”。
这些“药物”和“仪式”的功效,在一代代人的口口相传中得到强化,然后被记载到诸如《本草纲目》、《千金方》等典籍中。
于是我们才能在这些典籍中见识到“人中黄”、“金汁”、“紫河车”,甚至是“寡妇床头绳”这些荒谬的“药物”。
这就是中药问题上的“幸存者偏差”。
然后说“安慰剂效应”
安慰剂效应,指病人虽然获得无效的治疗,但却“预料”或“相信”治疗有效,而让病患症状得到舒缓的现象。
这是一种强大的心理暗示效应,据统计,安慰剂效应在普通人群中的发生率大约为20-25%。而在病人中安慰剂效应更容易出现,大约有35%的躯体疾病病人和40%的精神病病人都会出现此种效应。
也就是说,无论病人吃的是什么药,甚至不是药,只要他内心认为药物有效,安慰剂效应就有概率发生。
于是乎,安慰剂效应让很多本来毫无药用价值的普通植物、动物、矿物成了“药物”,而且,越是稀有的东西,安慰剂效应就越强,例如天山雪莲、千年人参、人形何首乌、虎骨熊胆,等等。
那么,怎么样才能避免安慰剂效应对于药物疗效的误导作用呢?
现代科学的办法是,进行双盲大样本随机实验。
什么是双盲大样本随机实验?
简单说,既然安慰剂效应会对药物实验的结果产生误导,那么在做实验的时候,就应该把受实验者分为两个对照组,在不告知对方的情况下,随机让一组服用安慰剂,另外一组服用参与临床试验的药物,然后对比实验结果,就可以清楚,该药物是真有疗效,还是某种安慰剂。
为什么要大样本呢?因为如果样本较小,结果偶然性太大,甚至可能存在作弊的情况。
如前所述,FDA的临床试验,第一期的主要目的是研究人对新药的耐受程度,了解新药在人体内的药代动力学过程。从第二期开始,除了了解药物的毒副反应,就要开始进行双盲实验,对药物的有效性进行基本评价。不过第二期的实验样本较小,容易出现偏差,所以进入第三期以后,才是真正的大样本阶段,可以真正对药物的安全性和有效性做出相对客观、全面的评估了。
这些由多个阶段组成的严格临床试验,让很多仅仅具有安慰剂效应的“传统药物”,露出了无能的真面目,也让一些具有长期毒性的“传统药物”,危害性一览无余。
在FDA要求临床试验阶段,上述那些中成药表现如何呢?
下面是来自2017年版《中国医药产业国际化蓝皮书》的统计结果:
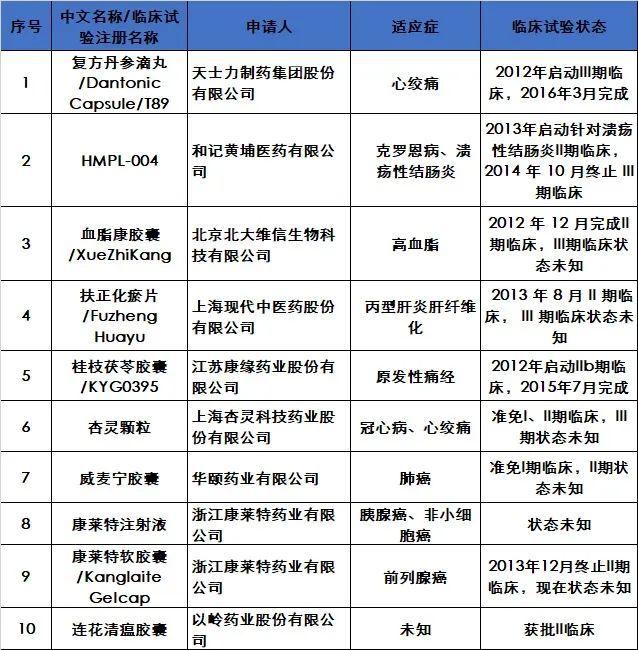
由上可知,截止2017年,上述药物所参加的临床试验,状态或为未知、或试验完成后没有进行下一步行动,结论是:没有一例中药,能够成功通过FDA三期临床试验。
那么从2017至今,这些药物在其主要适应症方面有什么进展呢?
通过查询美国临床试验注册登记网站(clinicaltrials.gov),我对上述中药企业在2017年之后向FDA提交的适应症试验结果(不包括适应症以外的研究)进行了一一查询。
复方丹参滴丸(T89)——天士力药业
2018年6月,天士力登记了一项T89预防及治疗急性高原反应的二期临床研究。该期研究于2019年11月结束。



2018年12月,天士力登记了一项研究:研究 T89 对稳定型心绞痛患者的抗心绞痛作用。

所登记的阶段是三期临床试验。

但试验尚未开始,目前仍处于人员招募阶段。

2020年2月,天士力登记了一项研究:T89 对改善 COVID-19 患者氧饱和度和临床症状的影响。

该试验目前已被取消。

2021年8月6日,天士力登记并启动了T89预防及治疗急性高原反应的研究

阶段为三期临床试验。

目前状态处于人员招募中。
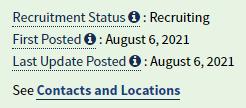
穿心莲制剂(HMPL-004)——和记黄埔医药
和记黄埔医药于2018年提交了一项关于HMPL004-6599的 I 期剂量递增研究,后因赞助商的原因,项目被终止了。
血脂康胶囊/XueZhiKang——北京北大维信生物科技有限公司
无进展。
扶正化瘀片(Fuzheng Huayu)——上海现代中药
未针对药物的主治功能进行有效性临床试验。
桂枝茯苓胶囊(KYG0395)——江苏康缘药业
无更新。
杏灵颗粒——上海杏灵药业
银杏药业于2017年提交了一项中草药治疗特发性肺纤维化临床试验,于2020年因招募不到足够符合条件的志愿者而终止了项目。

威麦宁胶囊——华颐药业
无更新。
康莱特注射液/软胶囊——浙江康莱特
2019年,浙江康莱特登记了康莱特注射液(KLTi)用于治疗晚期非小细胞肺癌(NSCLC)的临床试验,目前该研究处于人员招募阶段。

综上,自上世纪90年代以来,所有冲击FDA的中成药,至今无一款通过FDA的三期临床试验。
完










